Single cell WGS
Single-cell Whole Genome Sequencing
Motivation
The genome contained within each nucleus of an organism is exceedingly static, with an estimated frequency of mutation in somatic cells around 5x10-10 single nucleotide variants per bp per division. This rate of genomic change is bolstered by transposable elements and microsatellite instabilities1. However, cancers such as pancreatic ductal adenocarcinoma (PDAC) and triple negative breast cancer (TNBC), undergo marked genomic instability and may possess hypermutator phenotypes that can generate subclonal mutations during a tumor’s lifetime2. These phenotypes form when DNA repair mechanisms are disrupted. Single-cell whole genome sequencing is useful in these cases3. Heterogeneity in the tumor genome, such a copy number aberrations (CNAs, amplification or deletions of genomic regions) can be used to order the events of cancer progression, identifying prognostic markers and secondary mutations3. Darwinian selection can work within rapidly expanding neoplastic tumors, selecting for favorable mutations. Likewise, hypermutator phenotypes have been seen to be lost once cancer cells find local optima in fitness2. Additionally, single-cell analysis accounts for tumors with low cancer cell fraction or impure biopsy results, allowing for the distinction between tumor and unaffected somatic cells.
Method
Single-cell whole genome sequencing (scWGS) methods have three major criteria for success. They must capture the genome with high fidelity such that mutations can be faithfully called. They must have high coverage of the genome, in order to provide the statistical power to call copy number changes at high resolution. They must have unbiased coverage, such that there is a large signal-to-noise ratio. With these criteria in mind, the existing methods for scWGS are summarized below. Early protocols were focused on amplifying genomic DNA prior to library generation.
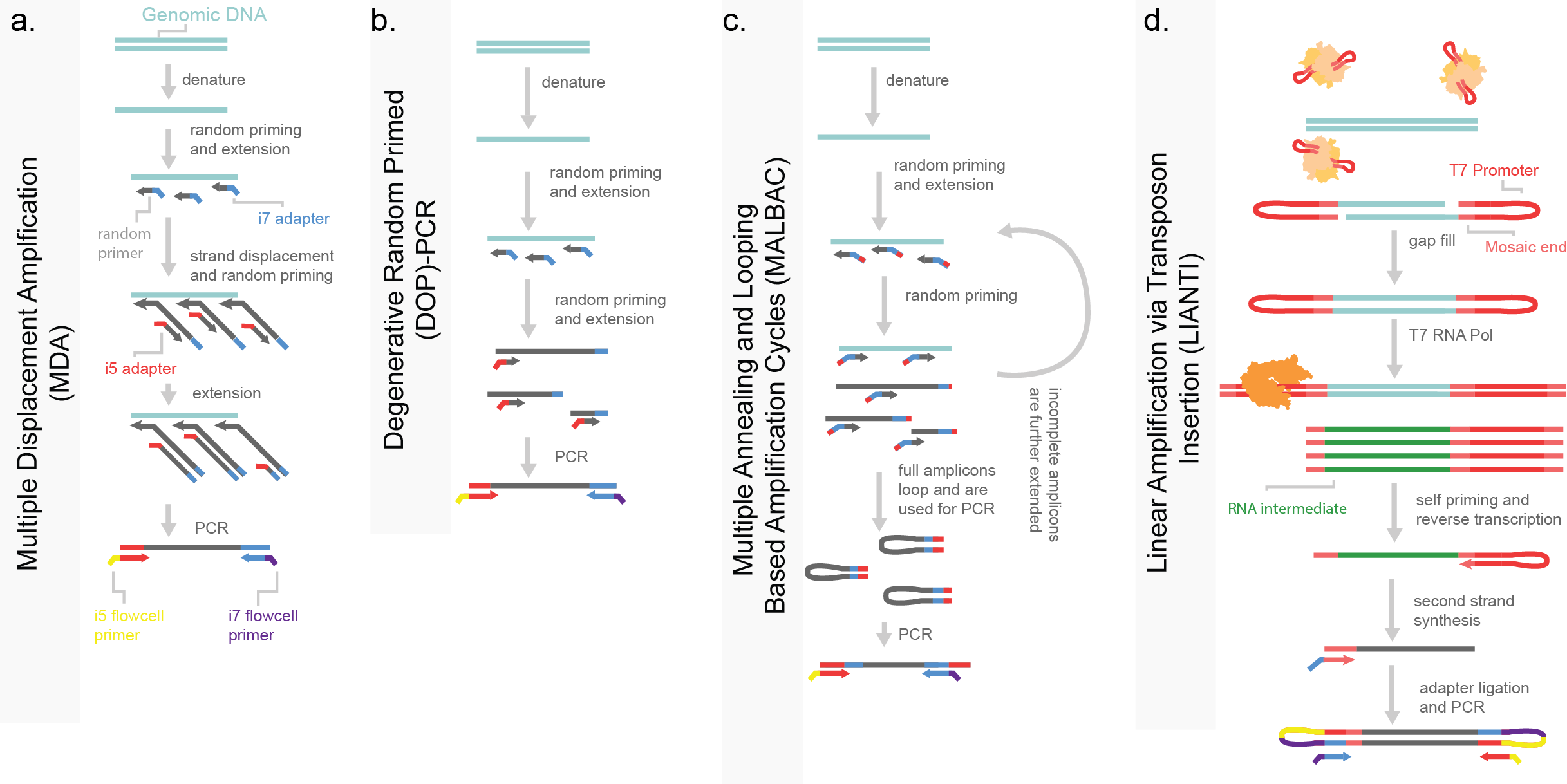
Figure 7. Pre-amplfication based single-cell whole genome sequencing (WGS). a) Multiple displacement amplification (MDA) is performed through the random priming across the whole genome, with subsequent random priming and amplification through a highly-processive polymerase. b) Degenerative random primed PCR, uses random priming to amplify the genome prior to PCR. c) Multiple annealing and looping based amplification cycles uses a random priming strategy with a specialized adapter which will self-hybridize to form intra-molecular hairpins. This self-annealing sequesters these molecules from further amplification. These hairpins are then PCR amplified. d) Linear amplification via transposon insertion (LIANTI) uses a Tn5 enzyme (shown in tan) to tagment DNA and insert a T7 promoter. This promoter is then used to in vitro transcription with an RNA T4 polymerase (shown in orange) to amplify the genome through RNA intermediates, which are then processed by reverse transcription, second strand synthesis and adapter ligation prior to PCR. Molecule coloring is consistent across panels.
Multiple-displacement amplification (MDA) uses random priming via degenerative nucleotides in an attempt to capture the genome in an unbiased fashion (Figure 7a). Amplicons captured across the genome are further amplified by the use of a highly processive polymerase like phi-29 with the generation of 1-2 μg of DNA (far exceeding the 6 pg contained within a cell)4. Despite the exceptionally high coverage, MDA produces significant biases due to the multiple rounds of PCR, making the detection of small CNAs difficult. Early examples of MDA were used to support a hypothesis of punctuated evolution in tumor progression in triple-negative breast cancer. This was done by measuring both the cells within the primary tumor and a subsequent liver metastasis3. Degenerative oligonucleotide primed polymerase chain reaction amplification (DOP-PCR) is a similar attempt at a random-priming strategy, with limited run-away amplification (Figure 7b). However, this method suffers from low coverage and substantial dropout. A mixture between these two methods, MDA and DOP-PCR, emerged in which PCR amplicons self-sequester after amplification by formation of a thermodynamically stable intramolecular loop in a method named MALBAC (multiple annealing and looping based amplification cycles). This makes PCR amplification “quasilinear” rather than exponential and leads for high coverage and less capture bias. However, MALBAC is not without limitations, the enzyme used for amplification (Bst large fragment) is error prone and the multiple rounds of linear amplification limit cellular throughput and increase cost (Figure 7c)5. LIANTI (linear amplification via transposon insertion) uses a transposase to introduce a T7 bacterial promoter region across genomic DNA. The T7 promoter is used for in vitro transcription genome wide, increasing the amount of material that can be generated per cell (Figure 7d). The RNA intermediate is then captured and converted to DNA libraries to be sequenced. While this method matches MDA in the amount of library material generated per cell, it has the shared limitation of uneven coverage. Further, error prone transcription exacerbates library quality, lowering the ability to call exacerbating library quality is the error prone intermediate states which decrease fidelity to the genome. This method has been developed for sci-compatibility as well wherein indexes are incorporated with a Tn5 tagmentation step6.
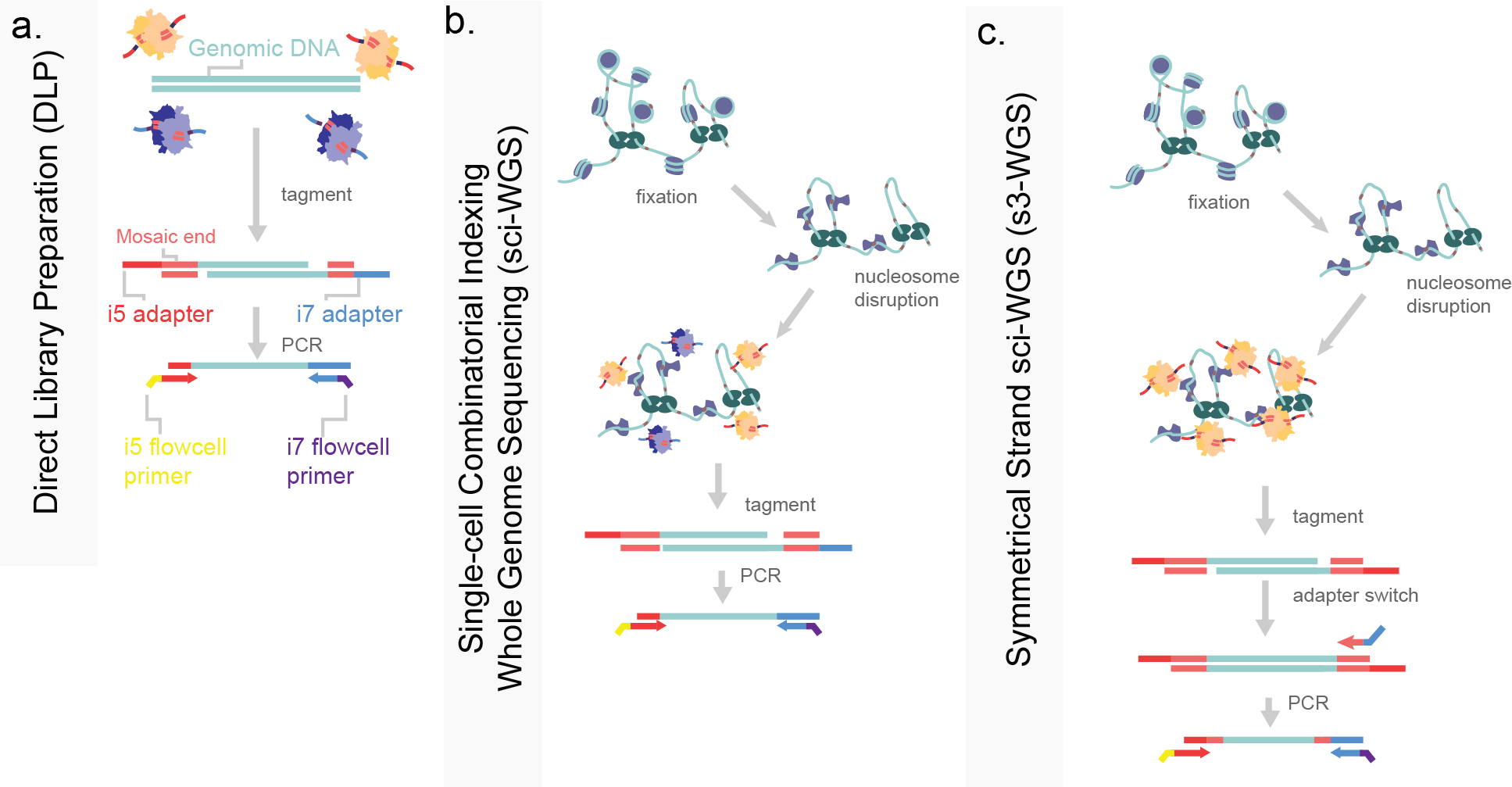
Figure 8. Tagmentation-based strategies of single-cell whole genome sequencing. a) Direct library preparation isolates single-cell genomic DNA in a well prior to full protein degradation. Purified genomic DNA is then tagmented with Tn5 enzymes loaded with i5 and i7 sequencing adapters (tan and purple respectively). b) Single-cell combinatorial indexing for WGS (sci-WGS) performs a fixation and nucleosome disruption in situ to render the genome accessible to tagmentation while maintaining nuclear integrity. The resulting tagmented DNA is then PCR amplified. c) Symmetrical strand sci-WGS uses similar pre-processing steps to sci-WGS, with the modification of Tn5 tagmentation such that library capture efficiency is higher.
Direct library preparation (DLP) avoids intermediate molecules by using Tn5 tagmentation to fragment the genome and introduce adapters for PCR, much like ATAC-seq (Figure 8a). In DLP, proteins associated with the genome are first denatured or digested prior to tagmentation, allowing for full accessibility. This method is simple and does not require pre-amplification, meaning a low rate of error introduction, and even coverage7,8. Evolutionary dynamics are prevalent in tumor samples, with known driver mutations often fixed in cancer cells. In parallel our group developed a similar method for higher cell count throughput. In this, we applied a similar approach to DLP using Tn5 for a sci-WGS. In order to denature genomic-associated proteins while maintaining the nuclear integrity needed for split-pool indexing, we used formaldehyde-based cross-linking and denatured with the detergent sodium dodecyl-sulfate (SDS; Figure 8b)9. The formaldehyde fixation maintained nuclear integrity while SDS denatured proteins, allowing for even tagmentation across the genome. I detail and adaptation to this strategy using the same s3- adapter switching strategy, increase genomic capture rate per cell to over 25% (Figure 8c). This increase in coverage allows for higher resolution assessment of copy number changes, and greater insight into which genes are driving cancer progression.
Analysis
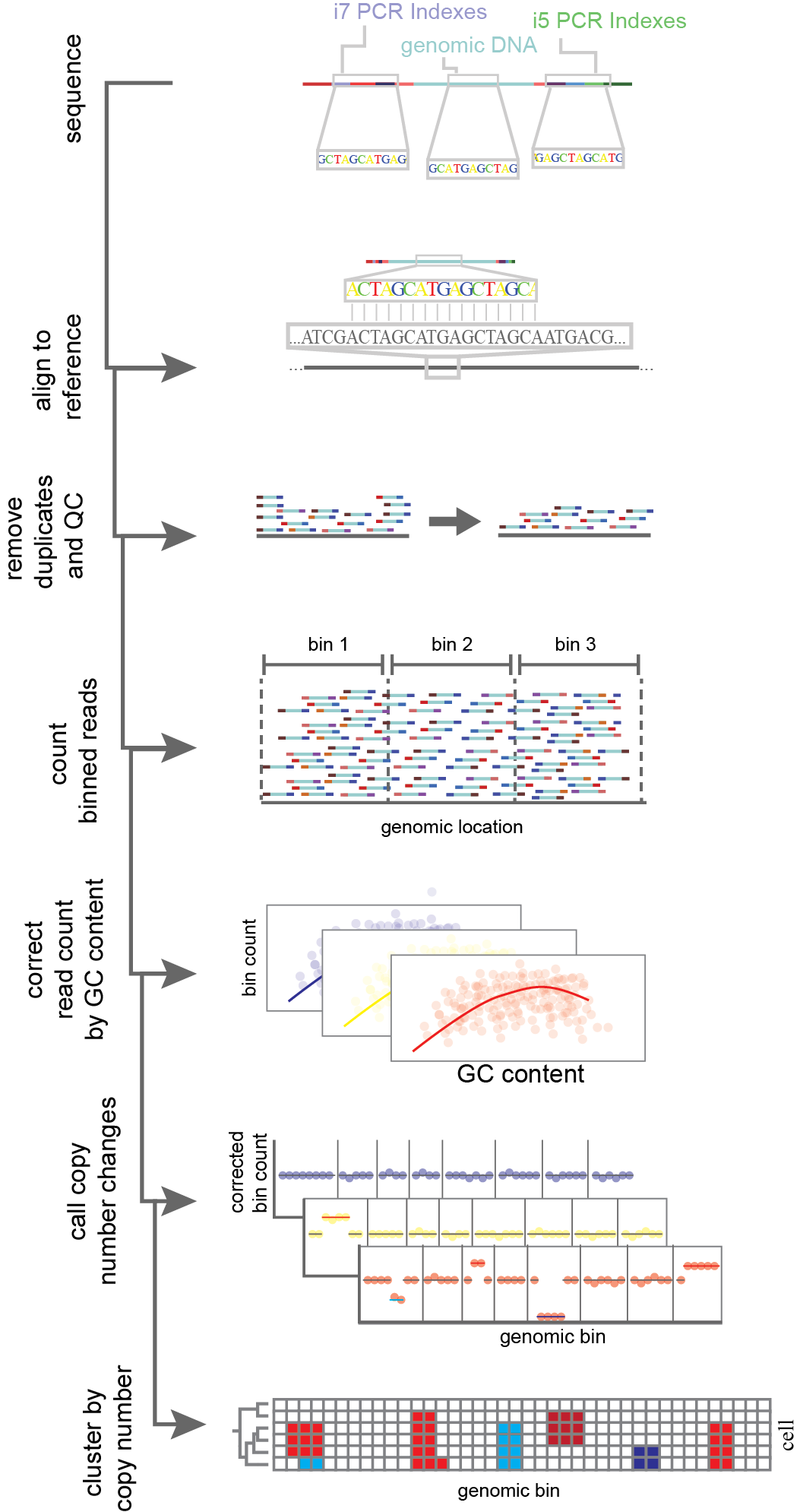
Figure 9. Analysis of copy number aberrations (CNAs) through single-cell whole genome sequencing. Reads are aligned to a reference genome after sequencing. Reads then undergo a quality control filter wherein low confidence mapping of reads and PCR duplicates are filtered out. For each cell, reads passing quality control are then counted within bins across the reference genome. Read count per bin in corrected for confounding factors such as GC content. After that normalization step, genomic bins undergo a segmentation where copy number changes are called based on changes in bin read count. Finally cells are grouped together based on shared CNAs to infer a lineage.
scWGS has the potential to capture both single-nucleotide variants (SNVs) between cell lineages as well as copy number aberrations (CNA). The read out for scWGS, just like scATAC, is count data. Reads captured are aligned to a reference genome (Figure 9). Depending on read count, the genome is then commonly segmented into “bins” or non-overlapping windows. Bins are used to aggregate data, allowing for enhanced statistical power in determining shifts in read counts, as well as to account for genomic biases. In the simplest form bins are a set length10. In other approaches bins are defined by a read count threshold, meaning each bin has the same number of reads by different lengths11,12. One common instance of genomic bias is the uneven dispersion of Guanine-Cytosine (GC) content. This is known to affect PCR efficiency and thus could bias results if left unaccounted. scWGS CNA callers work to normalize bins by one of two procedures. Either they use a set of cells known to be without CNAs to build a model for expected read counts per bin such as seen in SCOPE10, or they perform a normalization procedure to estimate ploidy. Normalization procedures include locally weighted linear regression (LOESS), or a modal regression followed by a post hoc means to estimate ploidy like in the tools Ginkgo11 and HMMcopy7. Following normalization across bins, the genome is then segmented to find where CNAs occur. Segmentation across bins occurs through either circular binary segmentation (CBS)10,11 or hidden Markov model (HMM)7. Both methods generate breakpoints, wherein bins shift from one state (e.g. diploid) to another (a deletion of amplification). Following this, cells can be hierarchically clustered to infer the phylogeny of CNAs. Further, SNVs can be attained per cell using variant-call tools, such as GATK13. From this data, haplotypes can be generated by shared changes. CNAs can be supported by observing shifts in the minor allele fraction; for instance if the minor allele fraction in a clonal population goes to 0, that supports a loss of heterozygosity event. Additionally clones can be hierarchically clustered by shared SNVs, similarly to CNAs7. In a breast cancer sample, Laks et al. uncovered a fixed amplification of oncogenes MYC, MCL1 and CCNE1, clonal loss of heterozygosity of BRCA2, and subclonal amplifications of RAD18 and RAB18. Subclonal alterations in tumor suppressors and oncogenes has the potential to inform precision medicine and our understanding of metastases7. Though single-cell methods are not necessary to detect these mutations, per se, the accuracy of population frequency and the co-occurrence of mutations within cells, provides a more confident picture of cancer progression.
References
- Blokzijl, F. et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature 538, 260–264 (2016).
- Loeb, L. A. A mutator phenotype in cancer. Cancer Res. 61, 3230–3239 (2001).
- Navin, N. et al. Tumour evolution inferred by single-cell sequencing. Nature 472, 90–95 (2011).
- Paez, J. G. et al. Genome coverage and sequence fidelity of phi29 polymerase-based multiple strand displacement whole genome amplification. Nucleic Acids Res. 32, e71 (2004).
- Zong, C., Lu, S., Chapman, A. R. & Xie, X. S. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science (80-. ). 338, 1622–1626 (2012).
- Yin, Y. et al. High-Throughput Single-Cell Sequencing with Linear Amplification. Mol. Cell 76, 676-690.e10 (2019).
- Laks, E. et al. Clonal Decomposition and DNA Replication States Defined by Scaled Single-Cell Genome Sequencing. Cell 179, 1207-1221.e22 (2019).
- Zahn, H. et al. Scalable whole-genome single-cell library preparation without preamplification. Nat. Methods 14, 167–173 (2017).
- Vitak, S. A. et al. Sequencing thousands of single-cell genomes with combinatorial indexing. Nat. Methods 14, 302–308 (2017).
- Wang, R., Lin, D. Y. & Jiang, Y. SCOPE: A Normalization and Copy-Number Estimation Method for Single-Cell DNA Sequencing. Cell Syst. 10, 445-452.e6 (2020).
- Garvin, T. et al. Interactive analysis and assessment of single-cell copy-number variations. Nature Methods 12, 1058–1060 (2015).
- Wang, X., Chen, H. & Zhang, N. R. DNA copy number profiling using single-cell sequencing. Brief. Bioinform. 19, 731–736 (2018).
- Poplin, R. et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 201178 (2017). doi:10.1101/201178